Search for articles:
Xian-Feng Sun , En-Hou Han
, En-Hou Han
Corresponding authors:
Received: 2018-03-29
Revised: 2018-05-25
Online: 2019-04-10
Copyright: 2019 Editorial board of Acta Metallurgica Sinica(English Letters) Copyright reserved, Editorial board of Acta Metallurgica Sinica(English Letters)
More
Abstract
The influence of Cr doping on the surface characteristics of Ni metal, including the surface energy, work function, adsorption and dissociation, has been investigated by means of first-principles calculation based on density functional theory. The results reveal that with the increase in Cr doping amount, the surface energies for all (111), (110) and (100) surfaces increase, whereas the work functions of (111) and (100) surfaces decrease followed by an increase and that of (110) surface has been decreasing. On the typical Ni(111) and Ni-Cr(111) surfaces, the adsorption energies follow the sequence O > OH > H > H2O, and the doping of Cr increases the chemical activity of surfaces leading to the improvement of adsorption energies. The H2O dissociating to OH and H is easier than the subsequent OH dissociating to O and H. The addition of Cr into Ni surface decreases the energy barriers and significantly increases the reaction rate constants.
Keywords:
Because of excellent mechanical properties and corrosion resistance, nickel-based alloys (such as 600, 690 and 800 alloys) are widely used in pressurized water reactor (PWR) and boiling water reactor (BWR) nuclear power plants as important structural components of primary loop, such as steam generator tube [1, 2, 3, 4]. The service environment of nickel-based alloy in nuclear power plant is very aggressive, with the increase in service time, pitting corrosion, intergranular corrosion and stress corrosion, etc., can attack these alloys aggressively, which seriously affect the safety and reliability of nuclear power plant [5, 6, 7].
The nature of the corrosion of nickel-based alloy in nuclear power plant is an electrochemical process which is tremendously relevant to surface characteristics, including the surface energy, work function, adsorption and dissociation characteristics. The surface energy and work function are two basic physical parameters on the metal surface [8, 9]. The surface energy reflects the stability of surface, whereas the work function suggests the difficulty of electrons to escape from the surface. They are of importance for the understanding and prediction of the crystal morphologies which can determine the subsequent reactions such as adsorption and dissociation [10]. Many investigations suggested that the surface energy and work function of pure Ni have a strong dependence on crystal orientation, and the Ni(111) surface is the most energetically favorable among the low index surfaces which is an evidence that the (111) surface has the largest possibility to be as surface exposed to vacuum [11], whereas the work function of (111) surface is the largest among the low index surfaces which indicates that it is more difficult for electrons to escape from (111) plane [12].
Furthermore, a serial of reactions involved in the process of corrosion are initialized by the adsorption of water molecule; subsequently water may dissociate into OH, O and H. The interaction between water with metal in aggressive environments leads to the dissolution of metallic atoms and the diffusion of elements. These processes are important in understanding the surface phenomenon, and extensive studies have been performed by theoretical methods to clarify the nature of the phenomenon. Xu et al. [13] investigated the adsorption of O atom on Ni(111) surface in a wide range of coverage by first-principles calculation. They found that the adsorption energy decreases with the coverage; on the contrary, the work function increases linearly with the coverage. It was concluded that the oxygen produces the dipole moment resulting in a large increase in the work function. Das et al. [14] used density functional theory to study the adsorption of atomic oxygen and water molecule on chromium-substituted Ni(111) surface. The results indicated that the net charge transfer from the Ni-Cr surface to the O atom is higher than that of Ni surface, and the hybridization between the metal d orbital and the oxygen p orbital occurs. The O bonding is mainly ionic, whereas the water bonds weakly on the surface. Additionally, Pozzo et al. [15] performed the investigation of water dissociation on Ni(111) surface using first-principles calculation. It was found that a barrier of 0.89 eV has to be overcome to dissociate the molecule into OH and H on the Ni(111) surface. Furthermore, the adsorption energy of dissociated products on Ni(111) is almost 0.5 eV lower than that of H2O, which means that the dissociation is thermodynamically advantageous.
It can be noted from the above introduction that for Ni-Cr binary alloy, only Das et al. performed some research work. However, their work just studied the adsorption of O and H2O on the surface of Ni-Cr alloy except OH and H. It is necessary to consider these four typical environmental particles more systematically. Furthermore, the surface energy and work function of Ni-Cr alloy and the dissociation of H2O on Ni-Cr surface have not been reported in the literature until now. These unclear aspects prevent us to have a deeper insight into the corrosion on the metal surface. It is generally believed that the element doping can change the geometry and electronic structures of system and the above processes extremely depend on the elements on the surface. The first-principles calculation based on density functional theory can obtain effective results from atomic scale perspective, whereas it is rather difficult to achieve through traditional experimental techniques. Therefore, the objective of this paper is to study the influence of Cr doping on the surface energy, work function, adsorption and dissociation. In the first section, we calculate the surface energy and work function of Ni surface with different Cr doping. Because the surface energy and work function have a strong dependence on the plane orientation, we have no idea what is the impact of crystal indices on surface energy and work function. Therefore, the three common surfaces of (111), (110) and (100) are selected to clarify this impact. Subsequently, the typical Ni(111) and Ni-Cr(111) surfaces are chosen to investigate the adsorption behaviors of O, OH, H and H2O as well as the dissociation process of H2O. Finally, the conclusions are summarized in the last section.
The calculation of density functional theory (DFT) was realized by VASP (Vienna ab initio simulation package) [16]. DFT is an accurate method to calculate the electronic structure, crystal energy and orbital interaction. It can help us have a better understanding of the fundamental phenomenon of the alloy surface and interface at an atomic scale. In this work, we selected projector-augmented plane wave (PAW) [17] as basic wave, using Perdew-Burke-Ernzerhof (PBE) [18] for the exchange correlation functions between electrons within the generalized gradient approximation (GGA) considering the first-order gradient of electron density. Compared with the local density approximation (LDA), GGA method can provide more accurate energy and structure.
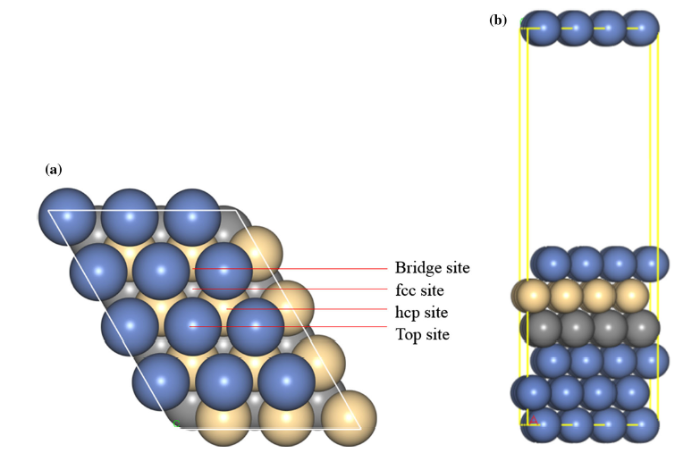
A periodic plate (slab) model was used to calculate the surface energy, work function, adsorption and dissociation, and the vacuum region thickness was 15 Å so that the interaction between different periodically repeated slabs can be neglected. Because the results showed that the errors in surface energy and work function were both less than 1% for six layers and nine layers on the Ni(111) surface, those on the Ni-Cr(111) surface were less than 2% and 0.5% so that the choosing of six layers was believed to be quite adequate to reach required accuracy. Therefore, the p(3 × 4) surface size and a six-layer model containing 72 atoms (each layer of the slab consists of 12 atoms) were used for (111), (110) and (100) surfaces. The middle two layer atoms were fixed for the calculation of surface energy and work function, whereas the bottom two layer atoms for adsorption and dissociation, and the other layer atoms were free to relax. The top and side views and the possible adsorption sites of the surface are shown in Fig. 1. We performed a k-point test, and the calculated energy difference between 6 × 6 × 2 and 3 × 3 × 1 k-points was less than 1% which indicates that 3 × 3 × 1 k-points are enough. In the geometry optimization, the convergence tolerance of energy and force was permitted up to 1.0 × 10-5 eV and 0.05 eV/Å, whereas for the self-consistent optimization, the criteria of energy convergence was increased to 1.0 × 10-6 eV. The cutoff energy of plane wave was set as 400 eV.
Fig. 1 a Top view, b side view of Ni(111) surface
The surface energy can be defined as the excess energy per unit area associated with the surface of a solid as compared with the bulk material [19]. The formula is as follows:
$\sigma = \frac{1}{2A}\left( {E_{\text{slab}} - nE_{\text{bulk}} } \right),$(1)
where Eslab is the total energy of slab layer, n is the total number of atoms, Ebulk is the energy of unit cell, and A is the surface area of slab layer.
The work function can be defined as the required energy of an electron in solid transferred far away from the surface [20]. In the thermal emission, the work function is the height of potential barrier that electrons need to stride across into vacuum region through quantum tunneling. Therefore, the metal with small work function is easier to lose electrons than that with larger work function with the following formula:
$\Phi = -e\varphi - E_{\text{f}} ,$(2)
where e is the charge of an electron, φ is the electrostatic potential in the vacuum nearby the surface. The term - eφ is the energy of an electron at rest in the vacuum nearby the surface. Ef is the Fermi level (electrochemical potential of electrons) inside the surface. The meaning of the term - Ef is the thermodynamic work required to remove an electron from the material to an infinite point of zero total energy.
The adsorption energy was calculated with the following formula:
$E_{\text{ad}} = E_{\text{slab}} + E_{\text{adsorbate}} - E_{\text{slab/adsorbate}} ,$(3)
where Eslab is the total energy of Ni or Ni-Cr surfaces, Eadsorbate is the energy of O/OH/H/H2O, and Eslab/adsorbate is the total energy of the adsorption system.
In this work, the reaction paths of water dissociation on the Ni and Ni-Cr surface were investigated by the nudged elastic band (NEB) method [21]. The transition-state search was performed by interpolating a series of images of the system between the initial and final states on the potential energy surface. Each image was optimized with the NEB algorithm. The highest point of the optimized reaction coordinate along the minimum energy path (MEP) should be the transition state along the chosen reaction path, and this highest energy relative to that of the initial state gave the activation barrier of the reaction.
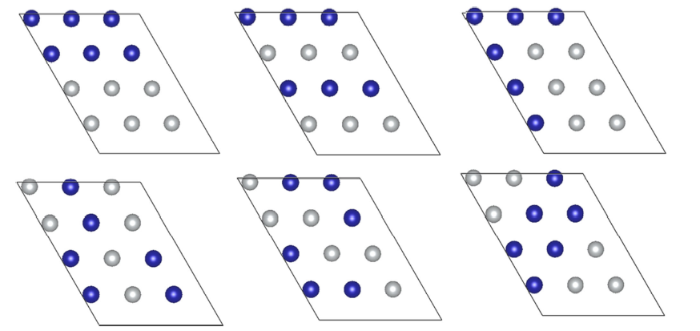
In this section, the effect of chromium doping amount in the Ni surface layer on the surface energy and work function was investigated. To clarity the impact of doping positions, we built six models that contain the same number of chromium atoms but with random atomic arrangements as shown in Fig. 2. The surface energies are 2.39, 2.42, 2.37, 2.32, 2.32 and 2.40 J/m2, respectively. The work functions are 5.10, 5.04, 4.90, 5.04, 4.92 and 4.99 eV, respectively. The errors in surface energy of different models are in the range of 4.1%, whereas the errors in work function are in the range of 2.0%. The differences in surface energy and work function among these models are very small, which indicates that the arrangement of Cr atom has little impact on the system calculations. Therefore, the Ni atoms are randomly replaced by Cr atoms in the next calculations.
Fig. 2 Arrangements of Cr in the Ni(111) surface layer. The blue one represents chromium and the gray one represents nickel
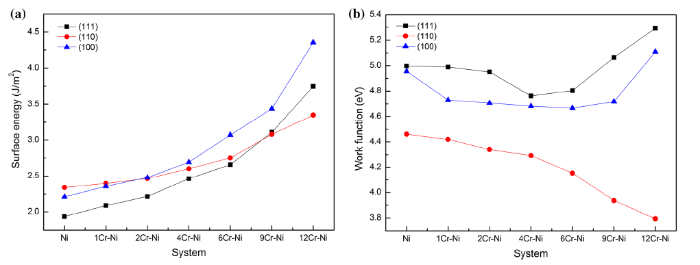
As shown in Fig. 3a, the surface energies of (111), (110) and (100) surfaces increase with the increase in Cr doping amount. This can be explained by the fact that the surface energy of Cr crystal is higher than that of Ni crystal; therefore, the surface energy of Ni-Cr binary alloy will increase as the doping amount increases. The changes in surface energies of (111) and (100) are confirmed to the identical law, whereas the surface energy of (110) changes significantly lower than those of the other two surfaces. At the beginning, the surface energies of (111), (100) and (110) are 1.94, 2.21, 2.34 J/m2, respectively, which are close to the previous calculation results of 2.01, 2.43 and 2.37 J/m2 [22]. Subsequently with the increase in Cr composition, the surface energies of (111) and (100) gradually exceed that of (110). Finally, when the Ni atoms in the top surface layer are completely substituted by Cr atoms (12 Cr atoms), the surface energy of (110) becomes the lowest among three surfaces, which is 3.35 J/m2, whereas the surface energy of (111) increases to 3.75 J/m2 and the surface energy of (100) becomes the largest, reaching 4.35 J/m2.
Fig. 3 Changes of a surface energies, b work function with the increase in Cr doping amount
Figure 3b reveals the relationship between work function and Cr doping amount. It can be observed that without Cr doping, the work functions of (111), (100) and (110) surfaces are 5.00, 4.96 and 4.46 eV, respectively, which are in good agreement with the literature values of 5.13, 4.93 and 4.57 eV [23]. Subsequently the work functions of (111) and (100) surfaces decrease followed by an increase with the increase in Cr doping amount. When the coverage of the surface is 1/3 (4 Cr atoms) and 1/2 (6 Cr atoms) for (111) and (100) surfaces, the work functions of the systems reach the lowest, which are 4.76 and 4.67 eV, respectively. However, the work function of (110) surface has been decreasing with the increase in Cr doping amount. When the surface layer are completely covered by Cr atoms, the work function of (110) surface has a minimum value of 3.79 eV, whereas those of (111) and (100) surfaces are increased to 5.29 and 5.11 eV, respectively. As is proposed by Leung et al. [24], he believed that the reasons leading to the change of work function are electrons transfer and polarization. It was found that the subtle change of charge transfer can determine the change of the sign and quantity of the surface dipole, which makes the work function strongly depend on the surface orientation. This theory can explain the relationship among charge transfer, surface dipole, polarization and work function, which can be described by
$\Delta \varphi = - \frac{e}{{\varepsilon_{0} }}[\Delta P_{1} + \Delta P_{2} ],$(4)
where e is the elementary charge and ε0 is the permittivity of vacuum, ΔP1 represents the change of the surface dipole density caused by charge transfer due to the difference in electronegativity, and ΔP2 represents the change of the surface dipole density caused by the polarization between doping layer and underlying substrate.
From above relationship, it is believed that the work function of a surface can be determined by the electronegativity of doping atoms and substrate atoms. The electrons in the doping layer can be attracted to the underlying substrate if the doping atom is less electronegative than the substrate atom. This can weaken the original surface dipole, which leads to a reduction in the work function. For the (110) surface, the change of the work function is governed by the electronegative difference between doped Cr atom and substrate Ni atom, which are 1.66 and 1.91, respectively. Because the doping layer is less electronegative than the underlying substrate, the work function has been decreasing as the doping amount increases. However, in the case of (111) and (100) surfaces, with the increase in Cr doping amount, the dipole changes due to polarization between doping layer and underlying substrate is gradually dominant, which can alter the sign of the surface dipole. This leads to an increase in the work function afterward.
From the above calculations, it can be seen that the (111) surface is the most stable on both Ni and Ni-Cr surfaces. Therefore, we selected the (111) surface and mainly considered the adsorption of four typical environmental particles, namely O atom, OH group, H atom and H2O molecule. They were placed at four kinds of adsorption sites, which are fcc hollow site, hcp hollow site, bridge site and top site, respectively, as shown in Fig. 1.
The calculated adsorption energies and metal-adsorbate distances are shown in Tables 1, 2, 3 and 4. It can be seen in Table 1 that for oxygen atom, the fcc site is the most preferential adsorption site with an adsorption energy of 5.76 eV on the Ni(111) surface, which agrees well with the previous literature values of 5.56 and 5.50 eV [14, 15, 25]. The hcp site is the next most stable site with an adsorption of 5.64 eV. Compared with the fcc site, the hcp site has no significant difference, only a little decrease. Because both fcc site and hcp site are 3-hollow sites [26], the oxygen atom can interact with three nearby metal atoms with a same bond length of 1.84 Å, almost consistent with the DFT p(2 × 2) result of 1.85 Å [14]. The adsorption energies of the bridge and top sites are 5.10 and 3.89 eV, respectively. Therefore, the top site is the most unstable site for oxygen adsorption. For Ni-Cr(111) surface, the adsorption energies increased to 6.58, 6.51, 6.30 and 6.13 eV for fcc, hcp, bridge and top sites, respectively. The increase in top adsorption is the largest which is up to 2.24 eV, whereas the fcc site only increases 0.82 eV. A previous DFT study [14] found that the O adsorption energy increases on the Ni-Cr(111) surface, which is the same as our calculation. Moreover, we note that the bond distances of Cr-O are shorter than those of Ni-O on the Ni-Cr(111) surface, which also suggests that the interaction between oxygen and chromium is stronger than that of nickel.
Table 1 Adsorption energies of oxygen as well as metal-oxygen bond distances on Ni(111) and Ni-Cr(111) surfaces
| Surface | Sites | This work Ead (eV) | Ni-O distance (Å) | Cr-O distance (Å) | Other studies Ead (eV) |
|---|---|---|---|---|---|
| Ni(111) | fcc | 5.76 | 1.84 | 5.56a 5.50b 5.56c | |
| hcp | 5.64 | 1.84 | 5.43a 5.41b 5.52c | ||
| Bridge | 5.10 | 1.79 | 5.28a 4.96b 5.08c | ||
| Top | 3.89 | 1.68 | 3.78a 3.69b 3.73c | ||
| Ni-Cr(111) | fcc | 6.58 | 1.90 | 1.76 | 6.31a |
| hcp | 6.51 | 1.90 | 1.76 | 6.24a | |
| Bridge | 6.30 | 1.80 | 1.70 | 6.05a | |
| Top | 6.13 | 1.60 | 5.88a |
Hydroxyl species are often formed on the metal surface [27]. The most energetically favorable adsorption site is fcc site for hydroxyl with an adsorption energy of 3.20 eV, as shown in Table 2. The OH is inclined to the normal of the surface, and the O atom is always at the bottom, which is consistent with the previous literature [29]. And the adsorption energies of hcp, bridge and top sites are 3.05, 2.79 and 2.00 eV, respectively. Similarly, the top site is also the most unstable. The adsorption energies of all sites for OH are lower than that of O, which indicates that the OH adsorption is weaker than that of atomic oxygen. The bond distances of nickel and oxygen for OH adsorption vary from 1.81 to 1.98 Å for all sites, whereas the O-H bond distances in OH groups are about 0.98 Å, which are close to the O-H bond length in H2O molecule, revealing that there is no dissociation of OH in the adsorption process. On the Cr doping surface, the adsorption energies have been increased to 4.02, 3.84, 3.44 and 3.48 eV, respectively, for fcc, hcp, bridge and top sites. And the increase in top adsorption is also the largest which is up to 1.48 eV, whereas the bridge site only increases 0.65 eV. The O-H bond distances in OH groups on the Ni-Cr(111) surface are the same as those on the Ni(111) surface, and the Cr-O distances on the Ni-Cr(111) surface are also shorter than those of Ni-O but longer than those of Cr-O for oxygen adsorption.
Table 2 Adsorption energies of hydroxyl as well as metal-oxygen bond distances on Ni(111) and Ni-Cr(111) surfaces
| Surface | Sites | This work Ead(eV) | Ni-O distance (Å) | Cr-O distance (Å) | Other studies Ead (eV) |
|---|---|---|---|---|---|
| Ni(111) | fcc | 3.20 | 1.97 | 3.07a | |
| hcp | 3.05 | 1.98 | 2.91a | ||
| Bridge | 2.79 | 1.92 | 2.93a | ||
| Top | 2.00 | 1.81 | |||
| Ni-Cr(111) | fcc | 4.02 | 2.02 | 1.93 | |
| hcp | 3.84 | 2.03 | 1.94 | ||
| Bridge | 3.44 | 1.99 | 1.88 | ||
| Top | 3.48 | 1.74 |
The adsorption energies of hydrogen are relatively lower than those of oxygen and hydroxyl adsorption, as shown in Table 3. They are 2.40, 2.40, 2.25 and 1.83 eV, respectively, for fcc, hcp, bridge and top sites, which indicates that the fcc and hcp sites are the most preferential, whereas the top site is the most unstable. For both fcc and hcp sites, the adsorption energies have no difference and in other ab initio calculations the energy difference between fcc and hcp sites can be almost neglected. The bond distances between hydrogen and three surrounding metal atoms for fcc and hcp sites are 1.70 and 1.71 Å, which is almost the same as the literature value of 1.71 Å [31]. The addition of Cr into Ni surface increases the adsorption energies and the fcc site makes the largest improvement to 2.90 eV, whereas the top site makes only a little improvement to 1.89 eV; meanwhile, the hydrogen atom is still closer to Cr atom than Ni.
Table 3 Adsorption energies of hydrogen as well as metal-hydrogen bond distances on Ni(111) and Ni-Cr(111) surfaces
| Surface | Sites | This work Ead (eV) | Ni-H distance (Å) | Cr-H distance (Å) | Other studies Ead (eV) |
|---|---|---|---|---|---|
| Ni(111) | fcc | 2.40 | 1.70 | 2.78a 2.69b | |
| hcp | 2.40 | 1.71 | 2.73a 2.69b | ||
| Bridge | 2.25 | 1.63 | 2.56b | ||
| Top | 1.83 | 1.47 | 2.33a 2.34b | ||
| Ni-Cr(111) | fcc | 2.90 | 1.89 | 1.68 | |
| hcp | 2.83 | 1.79 | 1.72 | ||
| Bridge | 2.54 | 1.75 | 1.75 | ||
| Top | 1.89 | 1.62 |
Table 4 Adsorption energies of water as well as metal-oxygen bond distances on Ni(111) and Ni-Cr(111) surfaces
| Surface | Sites | This work Ead (eV) | Ni-O distance (Å) | Cr-O distance (Å) | Other studies Ead (eV) |
|---|---|---|---|---|---|
| Ni(111) | Top | 0.31 | 2.15 | 0.52a 0.36b | |
| Ni-Cr(111) | Top | 0.79 | 2.10 | 0.80a |
For H2O molecule, it always migrates to top site in the calculation which demonstrates that the top site is the most energetically favorable adsorption site with an adsorption energy of 0.31 eV. Water is molecularly adsorbated on the surface through O atom and the orientation is almost parallel to the substrate. The distance between Ni and O atom in water is 2.15 Å, which is in good agreement in previous literature value of 2.14 Å [14]. Compared with the above adsorbates, the chemical bond strength between water and substrate is the weakest, which indicates that the surface metal atoms do not prefer to bond with water; therefore, H2O has a relatively high diffusivity on the surface. However, on the Ni-Cr(111) surface, the adsorption energy of water is up to 0.79 eV, which is two times more than that on the Ni(111) surface. As a matter of fact, the interaction between metal and water occurs due to the charge donation from the oxygen lone pair orbitals to the unoccupied p orbital, along with the admixture of this state with the metal d orbital, leading to p-d promotion [32].
From the above calculation results, it can be clearly observed that the adsorption energies of all adsorbates have been increased on the Ni-Cr(111) surface compared to those on the Ni(111) surface, which demonstrates that the Cr doping has a remarkable impact on surface chemical activity. The adsorption energies for both Ni(111) and Ni-Cr(111) surfaces follow the sequence O > OH > H > H2O. The most stable adsorption sites are fcc sites for O atom, OH group and H atom, whereas H2O molecule prefers to stay on the top site. The Cr doping does not change the most favorable sites. In order to further understand the interaction between metal and adsorbates, the Bader charge analysis is given in Table 5. Obviously, in the case of oxygen adsorption, the adsorbate gains the most electrons (0.834e) on the Ni(111) surface. It is an evidence that oxygen atom strongly interacts with the substrate. However, the electrons transferred from water molecule to metal atoms are the least (0.013e loss), which means a relatively weak bond with the surface. On the chromium-substituted surface, the gain and loss of electrons are increased to 0.874e, 0.541e, - 0.302e and - 0.038e, respectively, for O, OH, H and H2O adsorption. This demonstrates that the Cr doping can enhance the charge transfer between metal surface and adsorbates.
Table 5 Bader charge analysis for O, OH, H and H2O adsorption. The positive sign represents electron gain and negative sign represents electron loss
| System | Electron gain or loss of adsorbates | System | Electron gain or loss of adsorbates |
|---|---|---|---|
| Ni(111) + O | 0.834 | Ni-Cr(111) + O | 0.874 |
| Ni(111) + OH | 0.537 | Ni-Cr(111) + OH | 0.541 |
| Ni(111) + H | - 0.239 | Ni-Cr(111) + H | - 0.302 |
| Ni(111) + H2O | - 0.013 | Ni-Cr(111) + H2O | - 0.038 |
To study water dissociation, a number of MEPs have been analyzed to calculate the transition state. In order to be easier to compare the energies of the systems, the initial configurations are chosen to be zero datum. For H2O dissociation, the following elementary procedures on Ni(111) and Ni-Cr(111) surfaces are considered.
${\text{H}}_{2} {\text{O}}_{{ ( {\text{ads)}}}} \to {\text{OH}}_{{ ( {\text{ads)}}}} + {\text{H}}_{{ ( {\text{ads)}}}} ,$(5)
${\text{OH}}_{{ ( {\text{ads)}}}} + {\text{H}}_{{ ( {\text{ads)}}}} \to {\text{O}}_{{ ( {\text{ads)}}}} + 2{\text{H}}_{{ ( {\text{ads)}}}} .$(6)
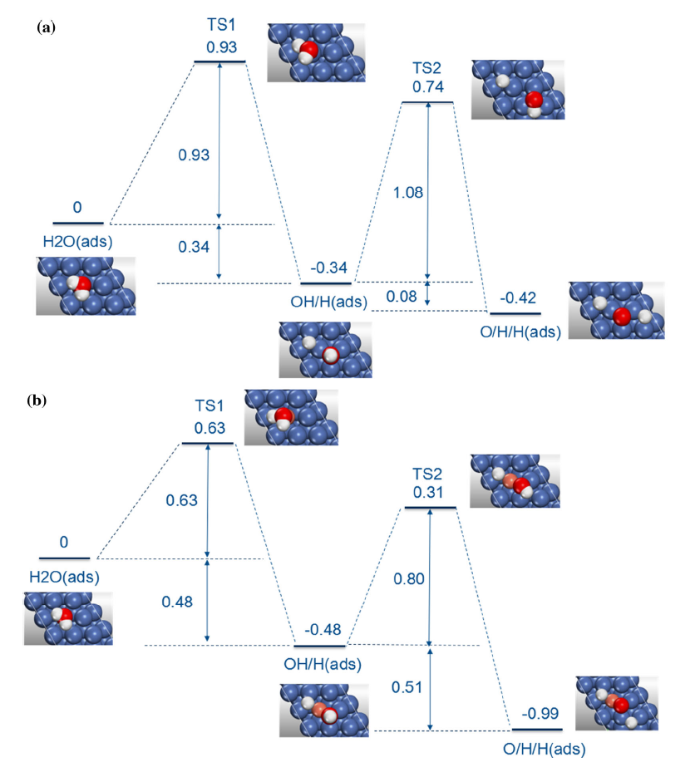
The initial and final states have been determined beforehand which are the most stable adsorption configurations of H2O, OH/H and O/H/H systems. Therefore, at first the water molecule is placed on the top site, developing to the OH/H adsorbed on the hollow sites, and at last two H atoms and O atom fall into the fcc sites. The transition states of the first and second dissociation step are shown in Fig. 4.
Fig. 4 Energy profile for dissociation of H2O on a Ni(111), b Ni-Cr(111) surfaces
For water molecule dissociation on the Ni(111) surface, four images are interpolated between initial and final states. At the initial state, the water molecule is placed almost parallel to the surface with a bond length of 0.98 Å in O-H and an angle of 106.1° between H2O planes. Then, the O atom in water moves closer to the surface, and its distance with the substrate becomes shorter reaching 1.93 Å at TS1 state (initial state: 2.15 Å). The distance between dissociated H and O atoms is 1.56 Å, which is longer than that at the initial state. At the final state of first dissociation, the dissociated H atom moves closer toward the three-hollow site and the OH falls into another fcc site nearby the original adsorption site which is almost vertical to the surface. The movement of OH and H leads to the decrease in the system energy. In the final stage, the energy of the system is - 0.34 eV which is lower than that of initial stage indicating that the OH/H co-adsorption is more stable. The negative sign shows that the reaction path is exothermic, and the energy barrier is 0.93 eV in the first dissociation (H2O → OH + H), which is in good agreement with the previous calculation result of 0.89 eV [15].
In the second step of dissociation (OH + H → O + 2H), the dissociated H has been always stayed at the initial site while the movement of OH is complicated. At the beginning, OH strides over bridge site falling into the nearby fcc site; meanwhile, the distance between O and H atoms becomes longer which is up to 1.35 Å. Then, O-H bond is broken and the newly dissociated H atom passes through bridge sites to search the most stable configuration. At last, the dissociated H and O atoms both locate at fcc sites with an energy of - 0.42 eV of the O/H/H system which is lower than that of OH/H system. From Fig. 4, it can be clearly seen that the energy barrier of the second step is 1.08 eV larger than that of first step indicating that the OH dissociation is more difficult than H2O dissociation.
To investigate the effect of Cr doping on the water dissociation, we repeat similar MEP calculations for the dissociation of H2O and OH on the Ni-Cr(111) surface. It is found that the process of water dissociation on Ni-Cr(111) surface is very similar with that on the Ni(111) surface. At the initial state, water molecularly locates at the top of Cr atom. In the TS1 and TS2 states, the bond lengths of the dissociated OH stretch 1.43 and 1.27 Å, respectively. Then the dissociated H atoms both move closer to the substrate. At the final state, the Cr-O distance is 1.76 Å shorter than that of Ni-O. Compared with Ni(111) surface, the energies of final states are much lower reaching - 0.48 and - 0.99 eV for OH/H and O/H/H, respectively, which demonstrates that the systems are more energetically preferable. The calculated reaction barriers of the first and second dissociation are 0.63 and 0.80 eV much lower than 0.93 and 1.08 eV, respectively, which can be explained by the increase in surface activity due to the addition of Cr making the O-H bond easier to break. From Bader charge analysis, we can see that the O atom in H2O molecule attracts 0.704e from H atom on the Ni(111) surface, whereas there are 0.664e electron transfers on the Ni-Cr(111) surface. This is because the O atom attracts more electrons from Cr atom and the transferred electrons between O atom and H atom decrease, which indicates that the O-H bond on the Ni-Cr(111) surface is weaker than that of Ni(111) surface.
According to Arrhenius equation k = A exp(- Ea/RT), it can be inferred that the reaction rate constants on the Ni-Cr(111) surface are 400 times and 300 times more than those on the Ni(111) surface for the first and second step of water dissociation when temperature is up to 300 °C. Therefore, the addition of Cr into Ni(111) surface dramatically increases the reaction rate of water dissociation. Subsequently, the oxide film on the metal surface is formed much easier to prevent further corrosion.
The influence of Cr doping on the surface energy, work function, adsorption and dissociation characteristics of Ni surface was investigated through density functional theory calculations. The model was represented by a six-layer slab with p(3 × 4) surface size, and a vacuum layer of 15 Å was employed to prevent the interaction between different periodically repeated slabs. The surface energies of (111), (110) and (100) surfaces increase with the Cr doping amount, whereas the work functions of (111) and (100) surfaces decrease followed by an increase and that of (110) surface has been decreasing. The arrangement of Cr atoms in the surface layer has little effect on the surface energy and work function. The adsorption energies of O atom, OH group, H atom and H2O molecule on the Ni-Cr(111) surface are higher than those on the Ni(111) surface. The Cr doping increases the chemical activity of the surface and makes the adsorbates bound with the substrate more easily. The most stable adsorption sites are fcc sites for O, OH and H, whereas H2O molecule prefers to stay on the top site. The Cr doping does not change the most favorable sites. The H2O dissociating to OH and H needs to overcome the energy barriers of 0.93 and 0.63 eV on the Ni(111) and Ni-Cr(111) surfaces, and the subsequent OH dissociating to O and H has the barriers of 1.08 and 0.80 eV. The presence of Cr weakens the O-H bond and decreases the reaction energy barrier.
Acknowledgements This work was supported by the National Key Research and Development Program of China (Grant No. 2017YFB0702100) in the Proof.
The authors have declared that no competing interests exist.

 WeChat
WeChat
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}