Search for articles:
Wei Diao
Corresponding authors:
Received: 2019-04-1
Revised: 2019-04-24
Online: 2019-12-10
Copyright: 2019 Editorial board of Acta Metallurgica Sinica(English Letters) Copyright reserved, Editorial board of Acta Metallurgica Sinica(English Letters)
More
Abstract
Microalloying is an effective approach to improve the mechanical properties of γ-TiAl intermetallic compound. Knowledge about the site occupancy of the ternary alloying element in the crystal lattice of γ-TiAl is highly demanded in order to understand the physics underlying the alloying effect. Previous first-principle methods-based thermodynamic models for the determination of the site occupancy were based on the point defect gas approximation with the interaction between the point defects neglected. In the present work, we include the point defect interaction energy in the thermodynamic model, which allows us to predict the site occupancy of the ternary alloying element in γ-TiAl beyond the point defect gas approximation. The model is applied to the γ-TiAl-Nb alloy. We show that, at low temperature, the site occupancy of Nb atoms depends on the composition of the alloy: Nb atoms occupy the Al sublattice for the Ti-rich alloy but occupy Ti sublattice for the Al-rich alloy. The fraction of Nb atoms occupying Al sublattice in the Ti-rich alloy decreases drastically, whereas the fraction of Nb atoms on the Ti sublattice in the Al-rich alloy decreases slightly with increasing temperature. At high temperature, Nb atoms occupy dominantly the Ti sublattice for both the Ti-rich and Al-rich alloys. The interaction between the point defects makes the Ti sublattice more favorable for the Nb atoms to occupy.
Keywords:
γ-TiAl intermetallic compound-based alloys have aroused considerable interests as potential candidates to manufacture turbine blades of jet engines and exhaust values of automobiles due to their excellent properties at elevated temperatures such as low density, high strength, good oxidation resistance and creep resistance [1]. However, because of their relatively low crystal symmetry (L10, base centered tetragonal with c/a > 1) and covalent nature of the Ti-Al bond due to the hybridization between Ti-3d2 and Al-3s23p3 electronic states, the material suffers poor ductility and workability at room temperature, which handicaps greatly the practical application of TiAl [2, 3]. Microalloying with ternary elements is an effective way to improve the mechanical properties of materials. For instances, V, Mn, Zr, Nb, Sc, Mo, Ni have been reported to enhance the plasticity of TiAl [4, 5, 6, 7, 8, 9]. 0.5 at.% C and Cr increase the strength and ductility of TiAl [10, 11, 12]. W and Y increase oxidation resistance of TiAl [13, 14]. C and Si enhance the creep properties of TiAl [15, 16]. Many efforts have been made to explore the physics of the alloying effect on the mechanical properties of TiAl. At electronic structure level, Morinaga et al. suggested that V, Cr and Mn substituting Al sites strengthen the d-d metallic bond and meanwhile weaken the covalence of Ti-Al interaction [17, 18], resulting in improved ductility of TiAl. At crystal lattice level, it has been proposed that the additions of V, Cr, Hf, Zr, Nb, Mn, etc., reduce the tetragonality of the lattice such that the possible deformation modes increase, leading to improvement in room-temperature ductility [19, 20, 21, 22]. At microstructure level, ternary additions such as V, Nb, Mo, Cr have been reported introducing cubic phases in γ-TiAl, which improves the high-temperature ductility and workability because the cubic phase provides more slip systems [23, 24, 25, 26, 27].
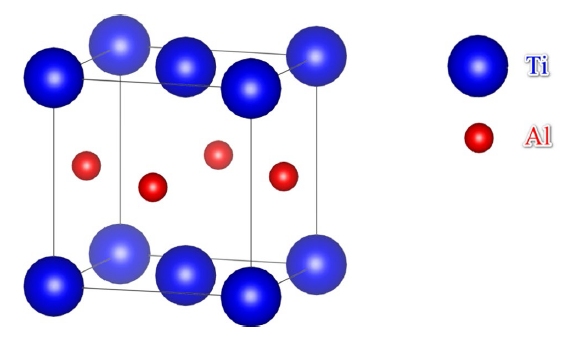
The crystal lattice of γ-TiAl consists of two sublattices with Ti and Al atoms occupying, respectively, one of them, see Fig. 1, for the unit cell of γ-TiAl. When taking different sublattices, the alloying atoms influence the atomic bond, crystal lattice, and phase stability in different ways. Therefore, it is crucial to know the site occupancy of the alloying atoms in γ-TiAl in order to understand the physics of the alloying effect on the mechanical properties. Various experimental techniques including atom location channeling enhanced microanalysis (ALCHEMI) [28, 29, 30], statistical ALCHEMI [31], X-ray scattering [32, 33], field-ion microscope (FIM) [34] and atom probe field-ion microscope (APFIM) [35] have been employed to determine the site occupancy of alloying atoms in γ-TiAl. The experimental measurements demonstrated that Nb [21, 28, 29, 30, 31, 32, 34, 35], Zr [21, 30, 31, 32], Hf [31] and Ta [31] occupy exclusively the Ti sublattice, whereas Fe [30, 33], Sn [30], W [31], Mo [31] and Ag [33] take the Al sublattice. The site occupancy of V, Cr and Mn is in dispute. Hao et al. [30] suggested that the site occupancy of V, Cr, Mn depends on the composition of TiAl, whereas other researchers reported exclusive site occupancy of them (Ti sublattice for V [29, 35] and Al sublattice for Cr [31] and Mn [21, 29, 32]).
Fig. 1 Unit cell of γ-TiAl with L10 structure
Theoretically, first-principles methods based on density functional theory (DFT) have been widely used to predict the site occupancy of ternary alloying atoms in γ-TiAl. In early times, the site occupancy was predicted simply by comparing some fundamental properties (indictors) of the systems with alloying atom occupying, respectively, Ti and Al sublattices. Xu et al. [36] took the bond order (a measure of the covalency of atomic bonds) from DV-Xα calculations as the indictor. The idea is that a higher bond order indicates a stronger covalent bond and the corresponding site occupation configuration is favored, noting that γ-TiAl exhibits partly covalent character. A better indicator may be the heat of formation that has been adopted by Erschbaumer et al. [37] and Wolf et al. [38] to determine the site occupancy of alloying atoms in γ-TiAl. A lower heat of formation corresponds to a more stable site occupation configuration. However, such predictions were made without considering the composition of the γ-TiAl-based alloy, whereas the site occupancy of some of the alloying elements may be composition dependent. On the other hand, the temperature effect on the site occupation was not taking into account.
Later on, thermodynamic statistical-mechanical models [39, 40, 41, 42, 43] were developed to describe the site occupancy of alloying element in TiAl. Woodward et al. [44] included the formation energies of the vacancies, antisites and the substitutional defects in the thermodynamic model, from which they predicted the site occupancy of substitutional ternary alloying atoms as the function of alloy composition and temperature. Jiang [45] further included the formation energy of the Wagner-Schottky defects, which allowed the author to determine more accurately the site occupancy at a given alloy composition and temperature. Adopting a three sublattice thermodynamic model, Wu et al. [46] predicted the site occupancy of 12 transition metal elements in γ-TiAl as functions of Ti/Al ratio and temperature. However, the above-mentioned thermodynamic models are based on the point defect gas approximation. Namely, the interactions between the point defects were absent in the model, and, therefore, it applies only to the dilute alloys. The influence of the interaction between the point defects in concentrated alloys on the site occupation remains to be explored.
In the present work, we aim to get some ideas on the influence of the point defect interaction on the site occupancy of ternary alloying atoms in γ-TiAl by developing a thermodynamic model beyond point defect gas approximation. The model is applied to the γ-TiAl with the addition of Nb. The fractions of Nb occupying Ti and Al sublattices are presented as functions of alloy composition and temperature.
The paper is arranged as follows. In Sect. 2, we describe the thermodynamic model for the prediction of the site occupancy and the first-principle calculations details. In Sect. 3, the formation energies of the point defects and the interaction energies between the point defects are presented, with which, the site occupancy of Nb in γ-TiAl is predicted. The obtained site occupancy is compared with available experimental measurements. We conclude our work in Sect. 4.
Let us consider an alloy A1-cBXc where A/B represents Ti/Al or Al/Ti depending on the ratio of Ti and Al in the alloy and X refers to Nb. Assuming that x amount of X occupies A sublattice (XA, direct site occupation) and y of X occupy B site (XB) while forming equal amount of BA antisite (indirect site occupation), we get x + y = c without considering the vacancies and intrinsic antisites. Then, the system may be expressed as (A1-x-yByXx)(B1-yXy). The corresponding Gibbs free energy at finite temperature T is
$G = E_{0} + xE_{\text{f}}^{{X_{A} }} + yE_{\text{f}}^{{X_{B} + B_{A} }} - TS,$ (1)
where E0 is the energy of the prefect TiAl. $E_{\text{f}}^{{X_{A} }}$ and $E_{\text{f}}^{{X_{B} + B_{A} }}$ are, respectively, the formation energies of an XA defect and an XB + BA defect pair. S is the mixing entropy:
$S = - k_{\text{B}} \left[ {\left( {1 - x - y} \right)\ln^{{\left( {1 - x - y} \right)}} + x\ln^{x} + y\ln^{y} + (1 - y)\ln^{(1 - y)} + y\ln^{y} } \right].$ (2)
At thermal equilibrium state, we have
$\frac{{{\text{d}}G}}{{{\text{d}}x}} = 0,$ (3)
with which we get
$\frac{{x\left( {1 - c + x} \right)}}{{(c - x)^{2} }} = \exp \left( { - \frac{{E_{\text{f}}^{{X_{A} }} - E_{\text{f}}^{{X_{B} + B_{A} }} }}{{k_{\text{B}} T}}} \right),$ (4)
By solving Eq. (4), we obtain the fractions of X on A (x/c) and B (y/c = 1 - x/c) sublattices at giving temperature T and composition c.
The formation energies of XA defect and a XB + BA defect pair may be calculated by using first-principles methods based on DFT with supercell model. The direct occupation of n X atoms in AN-nBNXn alloy (N is the number of A and B lattice sites in the supercell) results in the formation of n XA defects. The formation energy of an XA defects may be evaluated as
$E_{\text{f}}^{{X_{A} }} (n) = \frac{1}{n}\left[ {E_{{\left( {A_{N - n} X_{n} } \right)B_{N} }} - \left( {E_{{A_{N} B_{N} }} - n\mu_{A} + n\mu_{X} } \right)} \right],$ (5)
where $E_{{\left( {A_{N - n} X_{n} } \right)B_{N} }}$ is the total energy of AN-nBNXn supercell with n XA defects. $E_{{A_{N} B_{N} }}$ is the total energy of the unalloyed ANBN supercell. μA and μX are, respectively, the chemical potentials of A and X, which may be taking as the energies per atom of the elemental A and X. Similarly, the indirect occupation of n X atoms in AN-nBNXn alloy leads to the formation of n BA + XB defect pairs. The formation energy of an XB + BA defect pair may be expressed as
$E_{\text{f}}^{{X_{B} + B_{A} }} (n) = \frac{1}{n}\left[ {E_{{\left( {A_{N - n} B_{n} } \right)\left( {B_{N - n} X_{n} } \right)}} - \left( {E_{{A_{N} B_{N} }} - n\mu_{A} + n\mu_{X} } \right)} \right].$ (6)
If substituting one host atom by an alloying atom in a sufficiently large supercell (i.e., large N and n = 1), the induced defects are far from each other such that the interaction between them is weak and negligible. In this case, the above thermodynamic model corresponds to the one within the framework of the point defect gas approximation and is applicable only for dilute alloys. If the supercell contains more than one alloying atoms (n > 1), the induced point defects may interact with each other. The interaction is included implicitly in the formation energies of the point defect Eq. (5) and point defect pair Eq. (6). Therefore, the fraction x/c calculated using Eq. (4) with the formation energies $E_{\text{f}}^{{X_{A} }} (n)$ and $E_{\text{f}}^{{X_{B} + B_{A} }} (n)$ for n > 1 includes the contribution of the interaction between the point defects, which can be considered as beyond the point defect gas approximation. It should be noted that, in principle, when the interactions between the point defects and between the point defect pairs exist, the distribution of the point defects cannot be ideally random and the mixing entropy S expressed as Eq. (2) is not rigorous any more. However, in the present work, we still take Eq. (2) to evaluate the mixing entropy for simplicity. This approximation is similar to the one adopted to evaluate the free energy of regular solution [47].
In the present work, the size of the supercell is set as 3 × 3 × 3 times of the unit cell of γ-TiAl with L10 crystal structure, containing 54 Ti and 54 Al sublattice sites (N = 54). The distributions of the AX defects in the supercell with direct site occupation of the alloying atoms X and the AB + BX pairs in the supercell with indirect site occupation of X are described by using special quasi-random structure (SQS) scheme [48, 49]. The SQS supercells are constructed by using the Monte Carlo algorithm implemented by Walle et al. [50] in the alloy theoretic automatic toolkit (ATAT) package [51].
The first-principles plane-wave pseudopotential method based on density functional theory (DFT) [52] implemented in Vienna ab initio simulation package (VASP) is adopted to calculate the energies of the supercells [53, 54]. The projector augmented wave (PAW) potentials were used to describe the electron-core interaction [55, 56]. The semi-core 3s23p1 electrons of Al and 3s23p63d24s2 electrons of Ti and the semi-core 4s24p64d45s1 electrons of Nb were explicitly treated as valence. The exchange-correlation potential is described within the generalized gradient approximation parameterized by Perdew et al. (GGA-PBE) [57]. The k-point meshes for Brillouin zone sampling are constructed using the Monkhorst-Pack scheme [58], which is 5 × 5 × 5 for the supercells with N = 54. The plane-wave cutoff energy is set as 500 eV. The tolerance for the electronic structure minimization is set as 10-5 eV. The crystal lattices including the internal atomic positions of the supercells are fully relaxed by using the conjugate-gradient scheme. The Hellmann-Feynman force tolerance for the optimization of atomic positions was set to 0.01 eV/Å.
The lattice parameters, heat of formation, and bulk modulus of perfect γ-TiAl are listed in Table 1 in comparison with experimental and other theoretical results. Our calculated values are in good agreement with those from the experimental measurements and previous DFT calculations, which assures that our calculation details such as the potential, the exchange-correlation functional, and the plane-wave cutoff energy are reliable for the description of γ-TiAl system.
Table 1 Lattice parameters (a and c/a), heat of formation (Hf), and bulk modulus (B) of perfect γ-TiAl in comparison with available experimental and theoretical values
| References | a (Å) | c/a | Hf (eV/Å) | B (GPa) |
|---|---|---|---|---|
| This work | 3.993 | 1.020 | - 0.405 | 114.5 |
| DFT | ||||
| Tang et al. [59]. | 3.981 | 1.024 | - 0.403 | 114.2 |
| Ghosh et al. [60]. | 3.989 | 1.020 | - 0.406 | 113.7 |
| Ghosh and Asta [61] | 3.981 | 1.025 | - 0.412 | 112.1 |
| Zou and Fu [62] | 3.953 | 1.010 | - 0.420 | |
| Asta et al. [63] | 3.992 | 1.012 | - 0.437 | 128.0 |
| Yu et al. [64] | 3.995 | 1.020 | - 0.408 | |
| Fu et al. [65] | 4.001 | 1.012 | ||
| Music and Schneider [66] | 4.003 | 1.014 | - 0.401 | 112.0 |
| Shu et al. [67] | 4.006 | 1.012 | 111.0 | |
| EXP | ||||
| Duwez and Taylor [68] | 3.997 | 1.018 | ||
| Sridharan et al. [69] | 4.001 | 1.017 | ||
| He et al. [70] | - 0.435 | 109.8 | ||
| Tanaka [71] | 3.975 | 1.023 | 110.0 | |
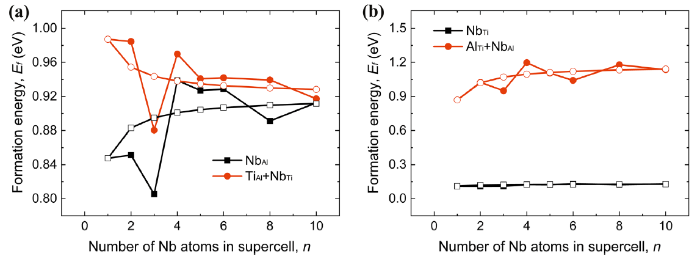
The formation energies of the point defect and point defect pair in both Ti-rich and Al-rich γ-TiAl are listed in Table 2 and also plotted in Fig. 2 as functions of the number of Nb atoms (n) in the N = 54 supercell.
Table 2 Formation energies of point defect and point defect pair in both Ti-rich and Al-rich γ-TiAl, calculated with different number n of Nb atoms in the supercell with N = 54
| n | Ti-rich (Ti54Al54-nNbn) | Al-rich (Ti54-nAl54Nbn) | ||
|---|---|---|---|---|
| NbAl | NbTi + TiAl | NbTi | NbAl + AlTi | |
| 1 | 0.848 | 0.987 | 0.110 | 0.869 |
| 2 | 0.851 | 0.985 | 0.109 | 1.024 |
| 3 | 0.805 | 0.880 | 0.111 | 0.951 |
| 4 | 0.939 | 0.970 | 0.125 | 1.198 |
| 5 | 0.927 | 0.941 | 0.123 | 1.105 |
| 6 | 0.929 | 0.942 | 0.131 | 1.041 |
| 8 | 0.891 | 0.939 | 0.124 | 1.179 |
| 10 | 0.913 | 0.917 | 0.130 | 1.136 |
Fig. 2 Formation energies of point defect and point defect pair as functions of the number of Nb atoms n containing in the supercells with, respectively, direct and indirect site occupations of Nb in Ti-rich a, Al-rich bγ-TiAl. The solid squares and circles are, respectively, for the calculated formation energies of the point defect in the supercell with direct site occupation of Nb and point defect pair in the supercell with indirect site occupation of Nb, $E_{\text{f}} \left( n \right)$. The opened ones represent the corresponding corrected formation energies, $\tilde{E}_{\text{f}} \left( n \right)$
As seen from Fig. 2a, for the Ti-rich Ti54Al54-nNbn alloy, the formation energy of the direct site occupation NbAl point defect is lower than that of the indirect site occupation TiAl + NbTi pair for all n, indicating that Nb atoms prefer the Al sublattice to the Ti sublattice at 0 K. For the Al-rich Ti54-nAl54Nbn alloy, the formation energy of the direct site occupation NbTi is lower than that of the indirect site occupation AlTi + NbAl pair, i.e., Nb atoms prefer the Ti sublattice to the Al sublattice at 0 K. The formation energy difference between the NbTi defect and AlTi + NbAl pair in the Al-rich alloy is much higher than that between the NbAl defect and TiAl + NbTi defect pair in the Ti-rich alloy. Namely, the above-mentioned site occupation preference of Nb is much stronger in the Al-rich alloy than in the Ti-rich alloy.
Roughly speaking, the calculated formation energy of NbAl point defect in the Ti-rich alloy (solid squares in Fig. 2a) increases, whereas that of the TiAl + NbTi defect pair (solid circles in Fig. 2a) decreases with increasing number of the Nb atoms n in the supercell. For the Al-rich alloys, the calculated formation energy of NbTi (solid squares in Fig. 2b) remains almost unchanged, whereas that of the AlTi + NbAl (solid circles in Fig. 2b) increases with n. However, the formation energies oscillate quite significantly against n. This is most likely due to the uncertainties associated with the distribution of the point defects in the supercell. In principle, one may find quite a few SQS supercells with different arrangements of the point defects for a given type of site occupation (direct or indirect) and alloy composition (here, n), leading to different interaction energies between the point defects. In order to obtain reliable formation energy, one should construct as many SQS supercells for a given type of site occupation and n, calculate the corresponding point defect formation energies, and then average them. However, this may be extremely time consuming and computation resource demanding. Instead, in the present work, we correct the formation energies as follows.
We first calculate the interaction energies between the point defect in the supercells containing different number of Nb atoms n with different types of site occupations. The interaction energy $\Delta E(n)$ between the point defects may be evaluated as
$\Delta E^{{{\text{Nb}}_{\text{Al}} }} (n) = \left[ {E_{{\left( {A_{N - n} X_{n} } \right)B_{N} }} + \left( {n - 1} \right)E_{{A_{N} B_{N} }} } \right] - nE_{{\left( {A_{N - 1} X} \right)B_{N} }} = n\left[ {E_{\text{f}}^{{X_{A} }} \left( n \right) - E_{\text{f}}^{{X_{A} }} \left( 1 \right)} \right],$ (7)
for the direct site occupation and
$\begin{aligned} \Delta E^{{{\text{Nb}}_{\text{Ti}} {\text{ + Ti}}_{\text{Al}} }} (n) & = \left[ {E_{{\left( {A_{N - n} B_{n} } \right)(B_{N - n} X_{n} )}} + (n - 1)E_{{A_{N} B_{N} }} } \right] - nE_{{\left( {A_{N - 1} B} \right)(B_{N - 1} X)}} \\ & = n\left[ {E_{\text{f}}^{{X_{B} + B_{A} }} \left( n \right) - E_{\text{f}}^{{X_{B} + B_{A} }} \left( 1 \right)} \right], \\ \end{aligned}$ (8)
for the indirect site occupation. Here, the N = 54 supercells containing n = 1 Nb atom are assumed to be large enough to avoid the interaction between the Nb atoms. Therefore, $E_{\text{f}}^{{X_{A} }} (1)$ and $E_{\text{f}}^{{X_{B} + B_{A} }} (1)$ contain no contributions from the interaction between Nb atoms and are treated as reference energies in Eqs. (7) and (8).
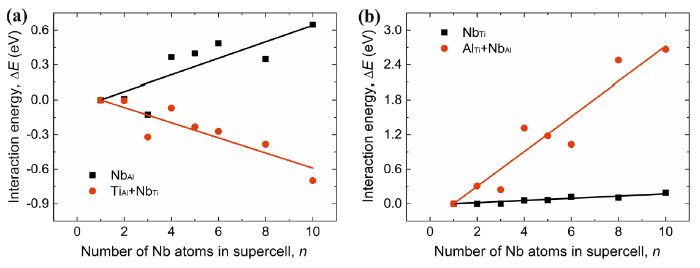
The interaction energies $\Delta E(n)$ are listed in Table 3 and plotted in Fig. 3. Roughly speaking, for the Ti-rich alloy (Fig. 3a), the interaction energy between the NbAl defects (Eq. 8), $\Delta E^{{{\text{Nb}}_{\text{Al}} }} (n)$, is positive and increases with increasing number of Nb atoms n in the supercells, indicating that the repulsion between the NbAl defects becomes stronger accordingly. On the other hand, the interaction energy between the NbTi + TiAl pairs (Eq. 9), $\Delta E^{{{\text{Nb}}_{\text{Ti}} + {\text{Ti}}_{\text{Al}} }} (n)$, is negative and decreases with increasing n, corresponding to stronger and stronger attraction between the NbTi + TiAl pairs. For the Al-rich alloy (Fig. 3b), the interaction energy between NbTi defects, $\Delta E^{{{\text{Nb}}_{\text{Ti}} }} (n)$, is positive with very small value and increases slightly with increasing n, indicating weak repulsion between the point defects. The interaction between the AlTi + NbAl pairs, $\Delta E^{{{\text{Nb}}_{\text{Al}} + {\text{Al}}_{\text{Ti}} }} (n)$, is repulsive and the interaction energy increases greatly with increasing n.
Table 3 Interaction energies between point defects in the supercells with direct and indirect site occupations of Nb for both Ti-rich and Al-rich γ-TiAl
| n | Ti-rich | Al-rich | ||
|---|---|---|---|---|
| Ti54Al54-nNbn | (Ti54-nNbn)(Al54-nTin) | (Ti54-nNbn)Al54 | (Ti54-nAln)(Al54-nNbn) | |
| 1 | 0 | 0 | 0 | 0 |
| 2 | 0.008 | - 0.005 | - 0.002 | 0.310 |
| 3 | - 0.127 | - 0.320 | 0.002 | 0.244 |
| 4 | 0.365 | - 0.070 | 0.059 | 1.314 |
| 5 | 0.397 | - 0.232 | 0.066 | 1.180 |
| 6 | 0.486 | - 0.271 | 0.122 | 1.033 |
| 8 | 0.349 | - 0.383 | 0.106 | 2.478 |
| 10 | 0.650 | - 0.699 | 0.193 | 2.665 |
| n | 0.071n - 0.071 | - 0.065n + 0.065 | 0.019n - 0.019 | 0.303n - 0.303 |
Fig. 3 Interaction energies between the point defects as functions of the number of Nb atoms n containing in the supercells with direct and indirect site occupations of Nb in Ti-rich a Al-rich bγ-TiAl. The solid squares and circles are, respectively, for the calculated interaction energies, $\Delta E(n)$, in the supercell with direct site occupation of Nb and in the supercell with indirect site occupation of Nb. The lines are for the corresponding interaction energies, $\Delta \tilde{E}(n)$, from the linear fitting of the calculated data points
As seen in Fig. 3, the calculated interaction energy $\Delta E(n)$ is somehow scattered against n due to the uncertainties associated with the specific arrangements of the point defects in the SQS supercells as discussed previously in this section. Despite the scattering of the data points, the calculated $\Delta E(n)$ follows roughly a linear trend against n. For each of the supercells with different n, the calculated $\Delta E(n)$ could be higher or lower than the averaged mean field value $\Delta \tilde{E}(n)$. Statistically, a linear fit of the calculated $\Delta E(n)$ against n is expected to represent the relationship between the averaged interaction energy $\Delta \tilde{E}(n)$ and n. It is noted that the slope and the intercept of the linear fit should be of the same value with opposite sign because the interaction energy for n = 1 is assumed to be 0. Namely, the fitting formula is ΔE(n) = an - a. The fitting equations are listed in Table 3 for both Ti-rich and Al-rich alloys with direct and indirect site occupation configurations.
Now, we may correct the formation energies of the point defects and point defect pairs according to Eqs. (7) and (8) by replacing the calculated interaction energy $\Delta E(n)$ with the fitted interaction energy $\Delta \tilde{E}(n)$. Namely,
$\tilde{E}_{\text{f}}^{{X_{A} }} (n) = \frac{1}{n}\Delta\tilde E^{{X_{A} }} (n) + E_{\text{f}}^{{X_{A} }} (1),$ (9)
$\tilde{E}_{\text{f}}^{{X_{B} + B_{A} }} (n) = \frac{1}{n}\Delta \tilde{E}^{{X_{B} + B_{A} }} (n) + E_{\text{f}}^{{X_{B} + B_{A} }} (1).$ (10)
The corrected formation energies $\tilde{E}_{\text{f}} \left( n \right)$ for both Ti-rich and Al-rich alloys with direct and indirect site occupations are also plotted in Fig. 2 as open squares and circles. It is shown that the calculated formation energies $E_{\text{f}} \left( n \right)$ oscillate around $\tilde{E}_{\text{f}} \left( n \right)$. The corrected formation energy $\tilde{E}_{\text{f}} \left( n \right)$ is expected to represent the average of the values from many SQS calculations. $E_{\text{f}}^{{X_{A} }} (1)$ and $\tilde{E}_{\text{f}}^{{X_{A} }} (1)$ are exactly the same because the N = 54 supercell containing n = 1 Nb atom is assumed to be large enough to avoid the interaction between the Nb atoms and treated as reference energy in the evaluation of the interaction energy (Eq. 7). This is also the case for $E_{\text{f}}^{{X_{B} + B_{A} }} (1)$ and $\tilde{E}_{\text{f}}^{{X_{B} + B_{A} }} (1)$. For all the point defects and point defect pairs, the formation energies $\tilde{E}_{\text{f}} \left( n \right)$ converge to stable values with n ≥ 8.
3.3.1 Point Defect Gas Approximation
With the formation energies of the XA point defect and XB + BA pair, the fraction of X occupying directly the A sublattice in the A1-cBXc alloy, x/c, may be calculated by using Eq. (3). As we mentioned previously in Sects. 2.1 and 3.2, $E_{\text{f}}^{{X_{A} }} (1)$ and $E_{\text{f}}^{{X_{B} + B_{A} }} (1)$ are assumed to be the formation energies for the point defect and point defect pair without interaction between the point defects and between the point defect pairs. Therefore, x/c evaluated with $E_{\text{f}}^{{X_{A} }} (1)$ and $E_{\text{f}}^{{X_{B} + B_{A} }} (1)$ corresponds to the fraction of X taking direct site occupation under point defect gas model. Figure 4 displays the fractions of Nb atoms taking direct site occupation configuration in both Ti-rich and Al-rich γ-TiAl within the framework of point defect gas approximation.
Fig. 4 Fraction (x/c) of Nb on Al sublattice in TiAl1-cNbc alloy a Nb on Ti sublattice in Ti1-cAlNbc alloy b within the framework of point defect gas model
For the Ti-rich TiAl1-cNbc alloy, the fraction x/c of NbAl is about 1 at temperature below about 100 K, i.e., the Nb atoms occupy solely the Al sublattice. With increasing temperature, the fraction x/c of NbAl decreases, indicating that Nb atoms transfer from the Al sublattice to the Ti sublattice to form NbTi + TiAl pair with equal fraction (1 - x/c) of NbTi and TiAl. It is expected that x/c converges to a fix finite value with increasing T as shown by the x/c-T curve for c = 1/54. Even at extremely high temperature, some of the Nb atoms are left on the Al sublattice with x/c > 0. With increasing Nb concentration c, the faction x/c of Nb left on the Al sublattice increases. For the Nb concentration c = 1/54, there is about 15% of Nb left on the Al sublattice at 1600 K, whereas for c = 9/54, the fraction goes up to about 38%.
For the Al-rich alloy, the Nb atoms occupy solely the Ti sublattice below about 700 K as the fraction x/c of NbTi is 1. Over about 700 K, a small fraction of the Nb atoms transfer from Ti sublattice to Al sublattice to form NbAl + AlTi pair as x/c for NbTi decreases whereas (1 - x/c) for NbAl and AlTi increases with increasing temperature. At the high-temperature end, x/c increases with increasing Nb concentration c, indicating more Nb atoms retain at the Ti sublattice and less transfer to Al sublattice.
3.3.2 Beyond Point Defect Gas Approximation
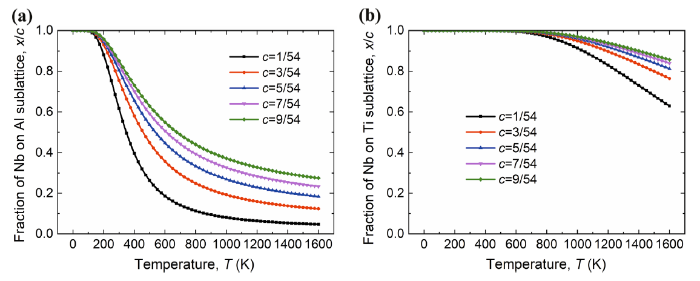
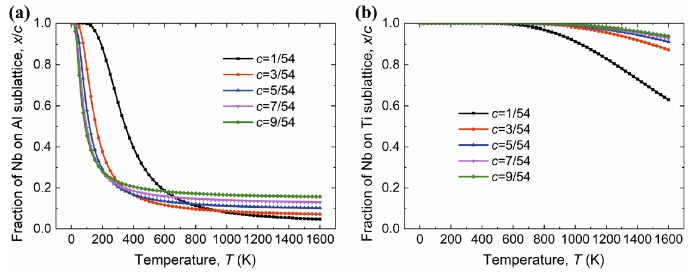
To consider the contribution of the interaction between the point defects on the site occupation of Nb in γ-TiAl, we take the corrected formation energy $\Delta \tilde{E}_{\text{f}} \left( n \right)$ [Eqs. (9) and (10)] to calculate the fraction (x/c) of Nb atoms taking direct site occupation according to Eq. (4). The calculated fractions x/c of Nb atoms occupying Al site (NbAl) in Ti-rich γ-TiAl and Nb atoms occupying Ti site (NbTi) in Al-rich γ-TiAl is displayed in Fig. 5 as functions of temperature T.
Fig. 5 Fraction (x/c) of Nb on Al sublattice in TiAl1-cNbc alloy a Nb on Ti sublattice in Ti1-cAlNbc alloy b taking into account of the interaction between the point defects
For the Ti-rich alloy, the fraction x/c of NbAl decreases with increasing temperature and converges to a fixed value when the temperature is high enough as shown in Fig. 5a, which is similar to the trend predicted within the framework of point defect gas approximation (Fig. 4a). However, at the low-temperature end, the fraction x/c of NbAl calculated with beyond point defect approximation decreases with increasing concentration c of Nb, in sharp contrast to that calculated with the point defect gas approximation. At the high-temperature end, x/c increases with increasing c but not as much as that predicted with the point defect gas approximation. For instance, at 1600 K, x/c for c = 9/54 is about 23% from beyond point defect gas model calculation, whereas it is 38% from point defect gas model calculation. The above results indicate that the interaction between the point defects makes the Ti sublattice more favorable for the Nb atoms to occupy.
For the Al-rich alloy, the trend of the fraction x/c of NbTi from calculations with beyond defect gas approximation is very close to that predicted with the point defect gas model. The only difference is that, at the high-temperature end, the decrease in x/c with increasing c is slower for the former than that for the latter. Namely, more Nb atoms occupy Ti sublattice if we go beyond the point defect approximation by taking into account the interaction between the point defects, i.e., the point defect interaction makes the Ti sublattice more favorable for the Nb atoms to occupy for the Al-rich alloy as well.
In Table 4, we list the site occupancy of Nb in γ-TiAl from various experimental measurements. It is seen that the Nb atoms occupy dominantly the Ti sublattice regardless of the composition of the alloy and heat treatment. This is in general agreement with our theoretical predictions at relatively high temperature. In different with other experiments showing that the Nb atoms occupying absolutely the Ti sublattice, the measurement of Mohandas et al. [29] demonstrated that there are about 11% of Nb left on Al sublattice for the Ti-54.0Al-2Nb alloy, which is supported by our calculations.
Table 4 Site occupancy of Nb in γ-TiAl from experimental measurements
| References | Method | Composition | Heat treatment | Site |
|---|---|---|---|---|
| Konitzer et al. [28] | ALCHEMI | Ti-47.0Al-5.0Nb, Ti-52.0Al-5.0Nb | Homogenized at 1000 °C, slow cooling to room temperature | Ti |
| Mohandas and Beaven [29] | ALCHEMI | Ti-54.0Al-2.0Nb | Homogenized at 1200 °C, equilibrated at 1300 °C, water quenched | 0.89Ti, 0.11Al |
| Hao et al. [30] | ALCHEMI | Ti-xAl-yNb, 46.0 < x < 53.0, 1.0 < y < 5.0 | Homogenized at 900 °C, ice water quenched | Ti |
| Rossouw et al. [31] | ALCHEMI | Ti-47.5Al-1.0Nb | Homogenized at 1300 °C | Ti |
| Doi et al. [32] | X-ray scattering | Ti-48.5Al-2.19Nb, Ti-47.69Al-5.52Nb | Homogenized at 1000 °C, water quenched | Ti |
| Kassab et al. [34] | FIM | Ti-48Al-5Nb | Levitation-melting in high vacuum | Ti |
| Wesemann et al. [35] | APFIM | Ti-45Al-5Nb | Homogenized at 1000 °C, cooled in air | Ti |
The occupation of Nb on the Al sublattice for the TiAl1-cNbc alloy at low temperature predicted by our calculations is not confirmed by the experiments because, in most experiments, the samples were heat treated at high temperature and then quenched such that the high-temperature site occupation retains to low temperature. Even the sample suffers from slow cooling might not reach the low-temperature equilibrium site occupation state because the site occupation transition occurs at relatively low temperature that does not allow the atomic diffusion.
In the present work, a thermodynamic model beyond the point defect gas approximation was developed to investigate the site occupancy of Nb atoms in γ-TiAl. The interaction energies between the point defects (NbAl and NbTi, respectively, in Ti- and Al-rich TiAl) and point defect pairs (TiAl + NbTi and AlTi + NbAl, respectively, in Ti- and Al-rich TiAl) were involved in the model. The predicted site occupancy is in agreement with experimental finding. The main results may be summarized as follows.
1. In the Ti-rich γ-TiAl-Nb alloy, the repulsion between the NbAl defects and the attraction between the TiAl + NbTi pairs become stronger with increasing Nb concentration. In the Al-rich alloy, the repulsion between the NbTi defects enhances slightly, whereas the repulsion between AlTi + NbAl pairs increases greatly with increasing Nb concentration.
2. At low temperature, Nb atoms occupy the Al sublattice for the Ti-rich alloy but occupy Ti sublattice for the Al-rich alloy. The fraction of NbAl in the Ti-rich alloy decreases drastically, whereas the fraction of NbTi in the Al-rich alloy decreases slightly with increasing temperature. At high temperature, Nb atoms occupy dominantly the Ti sublattice regardless of the composition of the alloy. The interaction between the point defects makes the Ti sublattice more favorable for the Nb atoms to occupy.
3. For the Ti-rich alloy, the fraction of NbAl decreases with increasing Nb concentration at low temperature, whereas the trend is opposite at high temperature. For the Al-rich alloy, the Nb concentration does not change the fraction of NbTi (almost 1) at low temperature but increases it at high temperature.
The authors are grateful for the financial supports from the National Key Research and Development Program of China under Grant No. 2016YFB0701301 and the National Natural Science Foundation of China under Grant No. 91860107.
The authors have declared that no competing interests exist.

 WeChat
WeChat
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}